Amyotrophic Lateral Sclerosis (ALS) is a fatal disease driven by motor-neuron atrophy, with no effective treatment or cure at present. Like many neurodegenerative diseases, defects in proteostasis occur; in particular, cytoplasmic mislocalization, accumulation and aggregation of an RNA-binding protein called TDP-43 is strongly implicated in ALS pathology for the vast majority of patients.
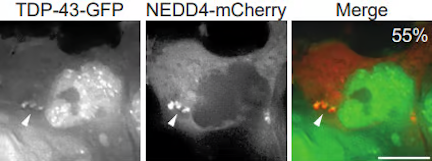
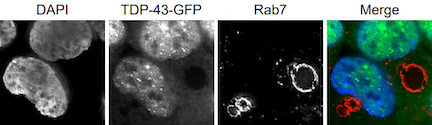
TDP-43 can localize in stress granules, and during our investigations of stress granule clearance mechanisms, we discovered a surprising new means of cytoplasmic TDP-43 degradation involving TDP-43 targeting to the endolysosomal pathway. We continue to work in this area to better understand mechanistically how TDP-43 degradation, including of so-called “toxic” species, can be upregulated, which may identify novel therapeutic targets in ALS.
More broadly, we are interested in what other cytoplasmic proteins degrade via this endolysosomal mechanism, and what governs the use of different degradation mechanisms for a given protein substrate.